文末更多GMP专题连载预告,先睹为快!
众所周知,细胞与基因疗法(CGT) 生产过程中使用的材料和试剂的品质对产品质量至关重要,尤其是外源因子污染等安全性方面控制引起越来越多的关注。外源因子通常是指无意中引入接种物、细胞基质和(或)生产制品所用的原材料及制品中的可复制或增殖的污染物,包括细菌、真菌、支原体和病毒等。
FDA也会建议使用可获得且可行的最高质量的材料和试剂用于细胞与基因疗法生产,这类材料和试剂常贴有“GMP级”标签,或有声称用于细胞治疗生产用途。一般来说,药物申报方应确保材料和试剂得到恰当的质量控制,以减轻给受试者或患者带来的不合理风险,如应该有某种方式来确保材料和试剂不含微生物和病毒污染物(无论是进行某种灭菌还是无菌测试)。对于CGT生产用原材料进行严格的质量控制,是降低CGT终产品中外源因子污染风险,保证CGT产品安全有效的必要措施,基于此,各国药典分别对CGT生产用原材料的安全性提出了相关的要求。
(1)FDA于2008年4月出台的《人类体细胞治疗新药临床试验申请的CMC药学信息的内容和审查》中的III 2 试剂 b.资格认定程序中要求如果该试剂未经FDA批准或通过,可能需要额外的测试来确保试剂的安全和质量。FDA建议研究者建立一个确认程序,包括安全性检测(无菌、内毒素、支原体和外源因子)、功能分析、纯度检测和测定(如残留溶剂检测),以证明不存在潜在有害物质。测试的程度将取决于在制造过程中如何具体使用特定的试剂。
(2)USP〈1043〉《细胞、基因和组织工程产品的辅助材料》中辅助材料(AM)的资格鉴定部分提到AM资格鉴定是获取和评估数据的过程,以确定生产工艺中使用的特定 AM 的来源、鉴别、纯度、安全性和整体适用性。
(3)EP 5.2.12 《用于细胞和基因治疗药物产品生产的生物来源的原料》中2,风险评估部分提到评价原料对细胞/基因治疗药物产品的质量、安全性和功效的影响必须由原料的使用者进行。任何单一的措施或措施的组合都不能保证原材料用于其预期用途的质量、功能和安全性。因此,风险评估必须考虑原材料的生物来源可追溯性、应用于原材料的生产步骤以及药品生产过程控制或从最终药品中去除原材料的能力。
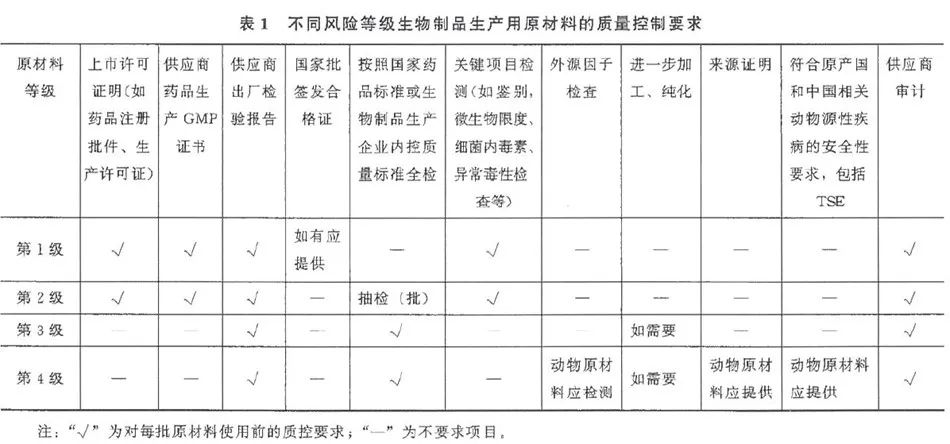
(4)中国药典三部《生物制品生产用原材料及辅料质量控制》中也写到生物制品生产工艺复杂且易受多种因素影响,生产过程中使用的各种材料来源复杂,可能引入外源因子或毒性化学材料。所以根据原材料的来源、生产以及对生物制品潜在的毒性和外源因子污染风险,可将生物制品生产用原材料风险级别从低到高分为以下4级,不同风险等级生物制品生产用原材料至少按表1进行质量控制。
ACROBiosystems百普赛斯GMP级别产品参考各国法规要求,对整个生产、质控过程中的外源因子污染等安全性进行了整体的设计和考量:
◉ 对经过驯化、建库后的生产用宿主细胞HEK293进行全面检定(26项)。
我们的GMP产品生产使用的宿主细胞HEK293参考如下法规进行检定,检测机构为国际知名第三方检测机构。<<<点击按钮,索取HEK293细胞COA
● ICH Q5第III部分细胞系检定:病毒测试;
● 1993 年的Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals (用于生产生物制品的细胞系检定中的考虑要点)(FDA)中V.QUALITY CONTROL TESTING部分要求;
● 2010 年的Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications(用于生产传染病适应症病毒疫苗的细胞基质和其他生物材料的检定和鉴定的行业指导方针)(FDA) ;
● 2020版中国药典三部《生物制品生产检定用动物细胞基质制备及质量控制》中(四)对于生产用细胞检定的基本要求。
◉ 宿主细胞经过构建后的工程细胞建库后再次进行检定,检测机构为国内知名第三方检测机构,检项有:无菌、支原体、细胞形态、体外不同指示细胞接种培养法、逆转录病毒、外源病毒检测等。
◉ 上游细胞培养阶段使用的培养基为化学成分明确培养基(Chemical Defined Medium,CDM),整个上游生产过程中所有物料均为无血清(Serum Free,SF)、无动物源性(Animal Origin Free,AOF)物料。细胞复苏及扩增阶段敞口操作均在C级洁净区的生物安全柜(Biosafety Cabinet,BSC)中进行,反应器生产阶段使用一次性密闭系统与无菌焊接技术,不同项目之间采用阶段式生产方式,从而极大地降低了发生污染和交叉污染风险。
◉ 下游纯化工艺设计之初,在纯化生产工艺中引入低pH和纳滤等病毒去除/灭活步骤,同时对于关键病毒去除步骤进行病毒去除工艺验证(进行中)。纯化过程使用的分离柱、层析介质等均为项目专用,且不用于不同的生产阶段,避免交叉污染风险。
病毒安全相关参考法规如下:
•《生物制品病毒安全性控制》,2020版药典,三部,生物制品通则
•《血液制品去除/灭活病毒技术方法及验证指导原则》国药监注[2002]160号
•《生物组织提取制品和真核细胞表达制品的病毒安全性评价技术审评一般原则》 CFDA编号:[S]GPH3-1,2005年12月
• Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. 1999. International Conference on Harmonization(ICH)Q5A(R1)
• Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products. EMEA /CHMP / BWP / 398498 / 2005. European Medicines Agency (EMA). 2009.
• Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal. USP General Chapters: <1050>.
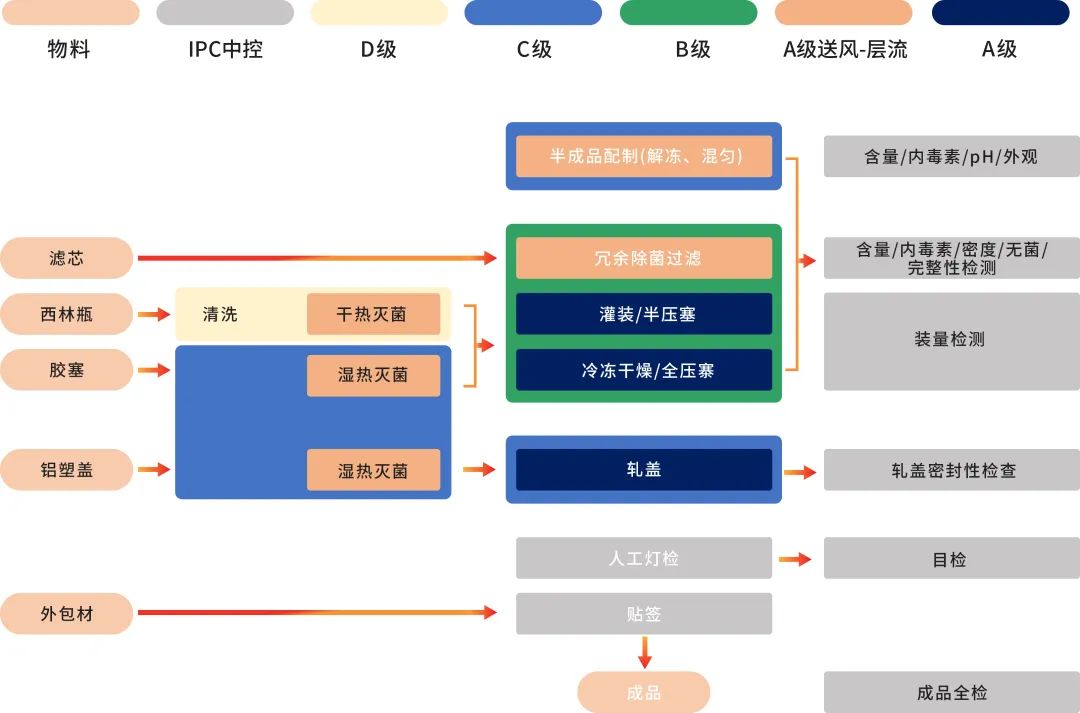
◉ 制剂生产在严格的B+A生产环境进行:原液经除菌过滤得到半成品后,使用全自动设备以及一次性无菌灌装系统,在B+A无菌环境下进行无菌部件组装、灌装、加塞、自动进出料和冻干等操作(PMS持续环境监测系统),冻干结束后在C+A环境下完成轧盖密封,获得的待包装产品经人工灯检、贴标等操作,并且检验合格后方可成品入库和放行。



















 Star Ribbon预染蛋白Marker蛋白质标记物是生物研究和药物开发的重要组成部分。无论是用于蛋白质电泳还是western blot,我们的预染色蛋白质标记物帮助您快速确定目标蛋白质的分子量或评估转移效率。Fc受体蛋白治疗性抗体的功效取决于Fab片段及其对目标抗原的结合活性,还取决于Fc片段及其与关键Fc受体的相互作用。因此,在抗体工程中候选物必须针对一系列受体进行测试。探索我们的重组Fc受体蛋白质的全面收藏!
Star Ribbon预染蛋白Marker蛋白质标记物是生物研究和药物开发的重要组成部分。无论是用于蛋白质电泳还是western blot,我们的预染色蛋白质标记物帮助您快速确定目标蛋白质的分子量或评估转移效率。Fc受体蛋白治疗性抗体的功效取决于Fab片段及其对目标抗原的结合活性,还取决于Fc片段及其与关键Fc受体的相互作用。因此,在抗体工程中候选物必须针对一系列受体进行测试。探索我们的重组Fc受体蛋白质的全面收藏!






























 膜杰作
膜杰作 Star Staining
Star Staining