Navigating TAM receptor dynamics in tumour immunotherapyYang, Chen, Wang
et alCancer Immunol Immunother (2025) 74 (5), 146
Abstract: The TAM receptor family is getting more and more attention in the field of tumour immunity. Activation of TAM receptors not only aids in the survival and multiplication of tumour cells but also increases their likelihood of invading other cells and spreading. In addition, activation of TAM receptors helps to inhibit the anti-tumour immune response, allowing tumour cells to evade immune surveillance. In terms of therapeutic strategies, a number of inhibitors targeting TAM receptors are in preclinical and clinical development. Despite significant progress in clinical trials in recent years, challenges remain. This review delves into the kinetic characteristics of the TAM receptor family, their dual role in tumour immunity, and the transmission process of downstream signalling pathways. Based on this, we analysed and summarised the unique strategies and combination therapies for regulating tumour immunity using TAM receptor inhibitors. It not only helps to elucidate the key role of TAM receptors in tumour immunity but also provides new perspectives and strategies for future tumour therapy.© 2025. The Author(s).
UNC9426, a Potent and Orally Bioavailable TYRO3-Specific InhibitorKong, Yang, Judd
et alJ Med Chem (2025)
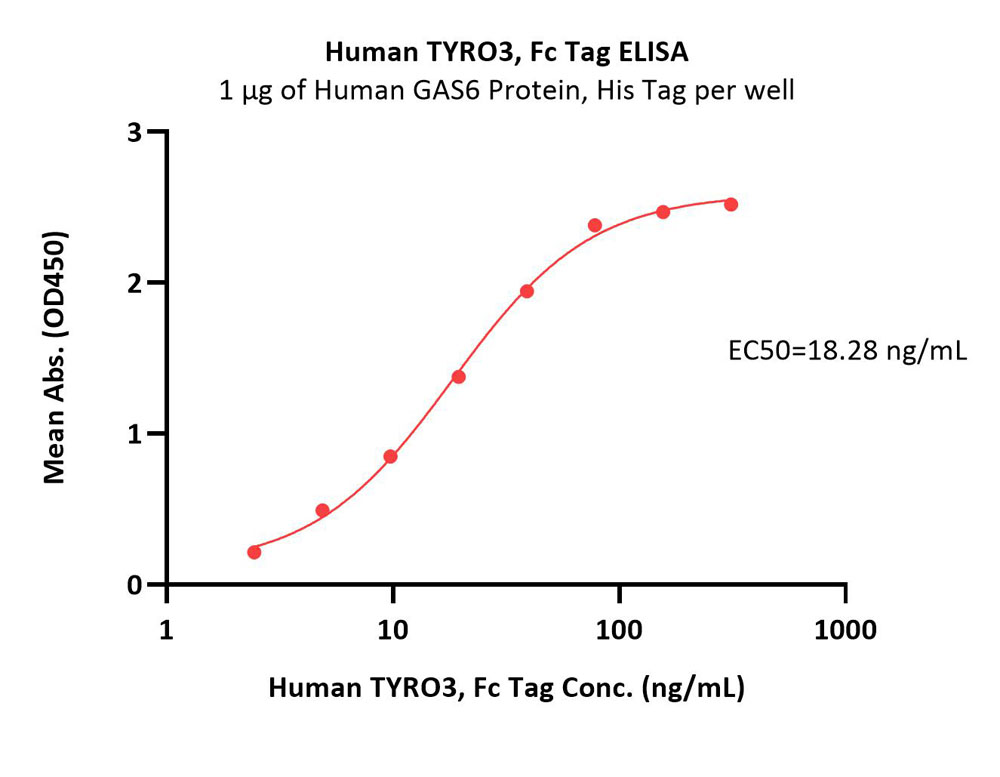
Abstract: TYRO3 plays a critical role in platelet aggregation as a platelet response amplifier. Selective inhibition of TYRO3 may provide therapeutic benefits for treating thrombosis and related diseases without increasing bleeding risk. We employed a structure-based approach and discovered a novel and potent TYRO3 inhibitor UNC9426 (12) with an excellent Ambit selectivity score (S50 (1.0 μM) = 0.026) and favorable pharmacokinetic properties in mice. Treatment with UNC9426 reduced platelet aggregation without increasing bleeding time and blocked TYRO3-dependent functions in tumor cells and macrophages, implicating its utility for multiple indications.
A single mutation in the PrM gene of Zika virus determines AXL dependency for infection of human neural cellsKhasa, Ogden, Wang
et alJ Virol (2025)
Abstract: Zika virus (ZIKV) is spread by mosquito bites and is unique among known flaviviruses for being able to cause microcephaly. Entry factors for ZIKV are incompletely understood, but phosphatidylserine (PS) receptors, including the TAM (Tyro3, AXL, and Mer) and TIM (T-cell Ig mucin) families, can serve as cofactors for flavivirus entry in a cell type-specific manner. We identify AXL as the top hit in a CRISPR/Cas9 genome-wide screen in human glioblastoma cells and establish a definitive role of AXL, but not TYRO3 or MerTK, for ZIKV infection. Additionally, Spondweni virus also shows AXL dependency, while dengue virus infection is not affected by AXL knockout. Passage of ZIKV in AXL knockout (KO) cells generated a mutant virus capable of infection via AXL-independent mechanisms, and multiple independent selections identified a common mutation, H83R, in the prM coding region of the ZIKV genome. The mutant virus exhibits an increased infectivity rate in AXL KO cells as compared to wild-type ZIKV and is dependent upon the single H83R mutation. The mutant virus' ability to infect cells in an AXL-independent manner is unrelated to interferon signaling antagonism but likely pertains to a change in virus maturation that leads to a structural disturbance of the ZIKV virion. Our study provides evidence for a potential mechanism linking the viral structural proteins and host PS receptor usage during flavivirus infection.IMPORTANCEA major challenge in elucidating the mechanism of Zika virus (ZIKV) pathogenesis is the multitude of cell types it infects with distinct requirements. The role of phosphatidylserine (PS) receptors in ZIKV infection is cell type-specific, and the controversy surrounds their function in flavivirus entry. Here, we establish a definitive requirement of AXL for infection of human glioblastoma cells by both Zika and Spondweni virus. We then identified a single amino acid mutation (H83R) in the prM protein of ZIKV that allowed AXL-independent infection of these cells. The H83R-mediated escape of AXL requirement is independent of interferon (IFN) signaling suppression by AXL; instead, the mutation has the potential to disrupt the virus assembly and virion structure. This study reveals a previously unknown connection between the PS receptor usage and the flavivirus prM gene, which can guide detailed molecular mechanism studies of the interplay between virion assembly and virus entry.
Proteolysis of TAM receptors in autoimmune diseases and cancer: what does it say to us?Malikova, Worth, Aliyeva
et alCell Death Dis (2025) 16 (1), 155
Abstract: Proteolytic processing of Receptor Tyrosine Kinases (RTKs) leads to the release of ectodomains in the extracellular space. These soluble ectodomains often retain the ligand binding activity and dampen canonical pathways by acting as decoy receptors. On the other hand, shedding the ectodomains may initiate new molecular events and diversification of signalling. Members of the TAM (TYRO3, AXL, MER) family of RTKs undergo proteolytic cleavage, and their soluble forms are present in the extracellular space and biological fluids. TAM receptors are expressed in professional phagocytes, mediating apoptotic cell clearance, and suppressing innate immunity. Enhanced shedding of TAM ectodomains is documented in autoimmune and some inflammatory conditions. Also, soluble TAM receptors are present at high levels in the biological fluids of cancer patients and are associated with poor survival. We outline the biology of TAM receptors and discuss how their proteolytic processing impacts autoimmunity and tumorigenesis. In autoimmune diseases, proteolysis of TAM receptors likely reflects reduced canonical signalling in professional phagocytes. In cancer, TAM receptors are expressed in the immune cells of the tumour microenvironment, where they control pathways facilitating immune evasion. In tumour cells, ectodomain shedding activates non-canonical TAM pathways, leading to epithelial-mesenchymal transition, metastasis, and drug resistance.© 2025. The Author(s).















































 膜杰作
膜杰作 Star Staining
Star Staining