JoGo-LILR caller: Unveiling and navigating the complex diversity of LILRB3-LILRA6 copy number haplotype structures with whole-genome sequencingNagasaki, Hirayasu, Khor
et alHum Immunol (2025) 86 (3), 111272
Abstract: Leukocyte immunoglobulin-like receptors (LILRs), encoded on human chromosome 19q13.4, comprise a set of 11 immunoglobulin superfamily receptors known for their genetic heterogeneity. Notably, LILRB3 and LILRA6 within this cluster exhibit pronounced sequence homology in immunoglobulin-like domains involved in ligand binding and variable copy number (CN) states. However, understanding their precise role remains challenging. To address this difficulty, we developed an algorithm and tool named JoGo-LILR Caller, which jointly calls CNs of LILRB3 and LILRA6 from a population-scale whole-genome short-read sequencing dataset. This tool was applied to 2,504 international HapMap samples and yielded a global CN profile. The 100 % concordance rate corroborated this profile with the CN data obtained from 40 samples of pangenome reference assemblies provided by the Human Pangenome Reference Consortium (HPRC). The frequencies of LILRB3-LILRA6 CN haplotype structures were also estimated for five continental groups with a global CN profile. The established allele frequency profile allowed our tool to estimate LILRB3-LILRA6 CN haplotype combinations. JoGo-LILR-trio enhanced the prediction reliability for haplotype pairs within trio datasets, with trio analysis on 40 child samples demonstrating a 100 % concordance between the predicted pair of haploid CN types and the diploid reference assemblies. Its utility will extend to facilitating software advancements for imputing LILRB3-LILRA6 CN types from SNP array genotyping data, enabling subsequent association analyses that link these CN types to diverse phenotypic traits and diseases, e.g., inflammatory bowel diseases and Takayasu arteritis.Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Identification of the hybrid gene LILRB5-3 by long-read sequencing and implication of its novel signaling functionHirayasu, Khor, Kawai
et alFront Immunol (2024) 15, 1398935
Abstract: Leukocyte immunoglobulin (Ig)-like receptors (LILRs) on human chromosome 19q13.4 encode 11 immunoglobulin superfamily receptors, exhibiting genetic diversity within and between human populations. Among the LILR genes, the genomic region surrounding LILRB3 and LILRA6 has yet to be fully characterized due to their significant sequence homology, which makes it difficult to differentiate between them. To examine the LILRB3 and LILRA6 genomic region, a tool named JoGo-LILR CN Caller, which can call copy number from short-read whole genome sequencing (srWGS) data, was applied to an extensive international srWGS dataset comprising 2,504 samples. During this process, a previously unreported loss of both LILRB3 and LILRA6 was detected in three samples. Using long-read sequencing of these samples, we have discovered a novel large deletion (33,692 bp) in the LILRB3 and LILRA6 genomic regions in the Japanese population. This deletion spanned three genes, LILRB3, LILRA6, and LILRB5, resulting in LILRB3 exons 12-13 being located immediately downstream of LILRB5 exons 1-12 with the loss of LILRA6, suggesting the potential expression of a hybrid gene between LILRB5 and LILRB3 (LILRB5-3). Transcription and subsequent translation of the LILRB5-3 hybrid gene were also verified. The hybrid junction was located within the intracellular domain, resulting in an LILRB5 extracellular domain fused to a partial LILRB3 intracellular domain with three immunoreceptor tyrosine-based inhibitory motifs (ITIMs), suggesting that LILRB5-3 acquired a novel signaling function. Further application of the JoGo-LILR tool to srWGS samples suggested the presence of the LILRB5-3 hybrid gene in the CEU population. Our findings provide insight into the genetic and functional diversity of the LILR family.Copyright © 2024 Hirayasu, Khor, Kawai, Shimada, Omae, Hasegawa, Hashikawa, Tanimoto, Ohashi, Hosomichi, Tajima, Nakamura, Nakamura, Tokunaga, Hanayama and Nagasaki.
Deciphering Immune-related Gene Signatures in Diabetic Retinopathy: Insights from In silico Analysis and In vitro ExperimentXia, Zhao, Xu
et alCurr Pharm Biotechnol (2024) 25 (15), 2032-2045
Abstract: Diabetes retinopathy (DR) is one of the most common microvascular consequences of diabetes, and the economic burden is increasing. Our aim is to decipher the relevant mechanisms of immune-related gene features in DR and explore biomarkers targeting DR. Provide a basis for the treatment and prevention of DR.The immune infiltration enrichment score of DR patients was evaluated from the single- cell RNA sequencing dataset, and the samples were divided into low immune subgroups and high immune subgroups based on this result. Through weighted gene correlation network analysis, differentially expressed genes (DEGs) between two subgroups were identified and crossed with genes with the strongest immune association, resulting in significant key genes. Then divide the DR individuals into two immune related differentially expressed gene (IDEG) clusters, A and B. Submit cross DEGs between two clusters through Gene Set Enrichment Analysis (GSEA) to further explore their functions. A protein-protein interaction (PPI) network of IDEG was established to further identify central genes associated with DR. Use the discovered central genes to predict the regulatory network involved in the pathogenesis of DR. Then, the role of the identified hub gene in the pathogenesis of DR was further studied through in vitro experiments.We found that the immune scores of DR and control groups were different, and 27 IDEGs were found in the DR subgroup. Compared with cluster A, the proportion of cytotoxic lymphocytes, B lineage, monocyte lineage, and fibroblasts in DR patients in cluster B is significantly enriched. GSEA indicates that these genes are associated with T cell activation, regulation of immune response processes, lymphocyte-mediated immunity, TNF signaling pathway, and other signaling pathways. The PPI network subsequently identified 10 hub genes in DR, including SIGLEC10, RGS10, PENK, FGD2, LILRA6, CIITA, EGR2, SIGLEC7, LILRB1, and CD300LB. The upstream regulatory network and lncRNA miRNA mRNA ceRNA network of these hub genes were ultimately constructed. The discovery and identification of these genes will provide biomarkers for targeted prediction and treatment of DR.By integrating bioinformatics analysis and in vitro experiments, we have identified a set of central genes, indicating that these genes can serve as potential biomarkers for DR, which may be promising targets for future DR immunotherapy interventions.Copyright© Bentham Science Publishers; For any queries, please email at epub@benthamscience.net.





















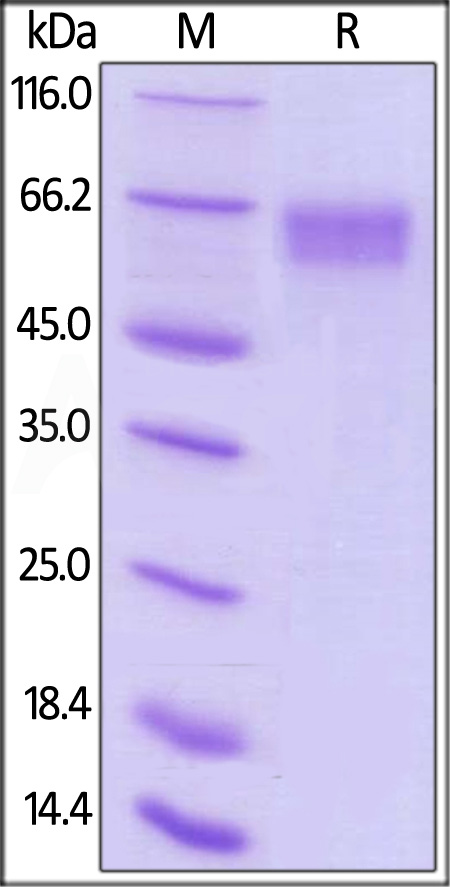
 Star Ribbon预染蛋白Marker蛋白质标记物是生物研究和药物开发的重要组成部分。无论是用于蛋白质电泳还是western blot,我们的预染色蛋白质标记物帮助您快速确定目标蛋白质的分子量或评估转移效率。Fc受体蛋白治疗性抗体的功效取决于Fab片段及其对目标抗原的结合活性,还取决于Fc片段及其与关键Fc受体的相互作用。因此,在抗体工程中候选物必须针对一系列受体进行测试。探索我们的重组Fc受体蛋白质的全面收藏!
Star Ribbon预染蛋白Marker蛋白质标记物是生物研究和药物开发的重要组成部分。无论是用于蛋白质电泳还是western blot,我们的预染色蛋白质标记物帮助您快速确定目标蛋白质的分子量或评估转移效率。Fc受体蛋白治疗性抗体的功效取决于Fab片段及其对目标抗原的结合活性,还取决于Fc片段及其与关键Fc受体的相互作用。因此,在抗体工程中候选物必须针对一系列受体进行测试。探索我们的重组Fc受体蛋白质的全面收藏!




























 膜杰作
膜杰作 Star Staining
Star Staining