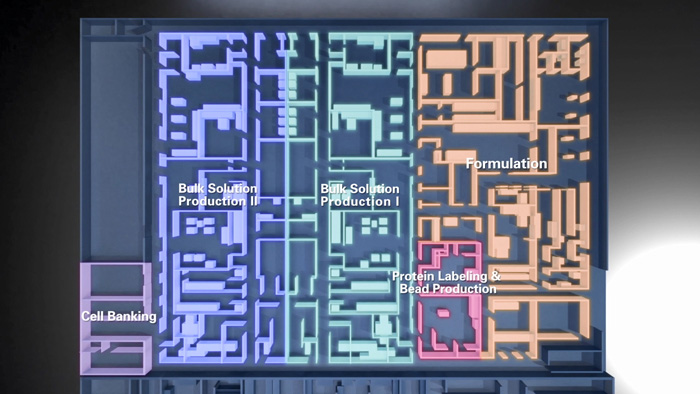
- 百普赛斯GMP工厂是百普赛斯集团全资子公司,于2024年6月正式投产。项目总投资3.5亿元。公司位于苏州市高新区,占地33亩 ,建筑总面积超过50,000㎡ 。采用智能化和模块化的设计,灵活韧性,能够胜任复杂多样的生产任务,满足客户GMP级核心原料的不同供应需求;工厂能够适应细胞治疗新技术的不断演进,通过创新实验室持续打造创新性的产品和解决方案,快速进行GMP级别产品升级和转化。GMP工厂是ADR(Audit-Ready)工厂,具备随时随地接受来自全球客户的线上线下审计能力。
- 质量体系遵循NMPA, US FDA, EMA等多国GMP法规,符合海内外申报要求;
- 产品包括GMP级细胞因子、抗体、酶、蛋白偶联磁珠、培养基等,助力CGT药物的研发、临床及商业化生产进程。
邀您开启一场专属虚拟之旅,一同走进百普赛斯GMP生产设施!在这里,您将深入探索先进的制造工艺,全面了解我们强大的GMP生产能力,还能与业内专家进行实时互动问答,获取最专业的解答与建议。深入洞察前沿的细胞与基因治疗(CGT)生产原料制造流程,挖掘潜在合作机遇,携手共创未来!
-
合理、科学的洁净车间设计:原液车间C+A,无菌制剂车间洁净级别B+A,符合药品级洁净度管控要求;
-
严格的APS无菌工艺验证:使用无菌营养培养基周期性模拟无菌生产工艺,并周期性确认人员无菌操作资质;
-
各区域空调系统独立控制,关键区域单向流控制,支持多个品种共线生产;
工艺用水遵循中国、美国和欧洲药典进行质量控制,并持续监测、确保水质可靠性;
-
符合FDA 21CFR Part 11要求的计算机化管理平台,从公用设施到文件系统,从电子时钟到数据自动备份等,提高工作效率的同时实现生产高自动化和产品生产质量可追溯性,并保障数据完整性;
-
经过验证的温度监控系统和仓储、物料管理系统(SAP系统)。独立的原材料待验、合格和不合格品区域,完整的物料管理体系和质量控制。
 研发和技术转移团队
研发和技术转移团队
- 5,000+重组蛋白成功开发经验
- 10+年蛋白质开发、生产和工艺放大经验
GMP生产团队
- 20+年生物行业生产管理经验
- 10+无菌制剂商业化生产经验
- 超过3000㎡GMP洁净车间
- 50+人生产团队
质量管理团队
- 超百人质量团队
- 近3000㎡QC实验室
- 团队拥有10+年药企质量管理经验
百普赛斯GMP工厂质量体系概述
GMP工厂按照NMPA, USFDA, EMA, ICH等各国GMP要求以及ISO9001和ISO13485标准建立质量管理体系。同时遵循国内外细胞和基因治疗药物产品生产用生物来源的原料管理指南,并且融入ICH风险管控理念,保障质量管理体系的持续高效运行。GMP工厂已获得ISO 9001和GMP双认证。质量管理人员拥有丰富的专业质量管理经验,熟悉全球不同国家地区生物医药相关法规,紧密追随行业趋势,可以根据不同产品类型、不同客户应用场景和不断变化的法规要求而快速建立或改进质量管理体系,进行相应的产品开发和生产,以满足不同客户的要求并使之与法规要求、市场环境等相适应。

探索我们的GMP品牌——Resilient Supply

针对制约细胞与基因治疗行业发展的诸如GMP核心原料质量波动、供应链脆弱及法规不确定性等难题,GMP品牌Resilient Supply,意在为全球客户带来更具灵活韧性、更低风险、更高品质且更专业支持的解决方案,加速CGT生物药研发上市进程。
Resilient Supply的内涵充分融入到了GMP工厂的设计和运营理念中。工厂采用智能化和模块化的设计,灵活韧性,能够胜任复杂多样的生产任务,满足客户GMP级核心原料的不同供应需求;工厂能够适应细胞治疗新技术的不断演进,通过创新实验室持续打造创新性的产品和解决方案,快速进行GMP级别产品升级和转化。GMP工厂的质量体系遵循中国、美国、欧洲等多国GMP法规和指南,是ADR(Audit-Ready)工厂,已具备随时随地接受来自全球客户的线上线下审计能力。
Resilient Supply核心优势, 开启 CGT 制造新视界!
| 细胞库/菌种库 |
过程控制 |
生产设施 |
• 无血清稳定株细胞株单克隆技术/无动物源大肠杆菌培养体系
• 分级建库,细胞库/菌种库权限管理,异地存储
• 原始宿主细胞全面检定(HEK293共检测28项;CHO共检测52项),无病毒等外源污染因子,符合按中美药典和ICH Q5
• 工程细胞全面检定,无病毒等外源污染因子,符合中美药典(无病毒等外源污染因子,共检测33项)
• 气相液氮罐存储降低交叉污染风险,在线温度持续监测、液氮液位监测及自动补充
• 建库环境为C+A,人员无菌操作资质
• 细胞库稳定性验证(遗传稳定性,传代稳定性,长期稳定性)
|
• 基于质量源于设计(QbD)的理念,研发阶段输出CQA、CPP及IPC策略,覆盖产品安全性、有效性
• 上游使用20L~500L生物反应器,PAT技术自动监测温度、溶氧、pH等关键参数
• 真核产品下游增加特定病毒灭活/去除步骤(1~2个),保证产品安全性
• 制剂灌装在线持续环境监测,无菌保障更可靠
• 制剂灌装阶段100%在线称重反馈,确保装量准确性
• 制剂阶段三批无菌工艺模拟验证/定期再验证(APS),保证无菌工艺的可靠性
• 制剂阶段二级冗余除菌过滤,过滤器使用前、后均进行完整性测试
• 工艺用水、气、洁净环境定期监测与回顾
|
• 设施设备持续验证及预防性维护管理,确保设施设备稳定运行
• 厂房设施多路供电模式,确保生产可持续性
• 工艺用水、工艺用气和洁净环境,符合国际GMP法规及ISO标准
• 关键功能间单向流设计,配置独立空调系统
• 独立细胞建库车间,避免交叉污染
• 一次性密闭生物反应系统,符合USP Class VI的要求
• 产品专用填料及层析柱自动CIP,避免交叉污染
• 料液穿墙管路输送,管路无菌焊接
• 洗瓶、烘干灭菌、灌装、轧盖全自动化操作,避免人为污染
• 严格B+A制剂生产环境,辅助区域A级延展层流送风保护
• 在线持续环境监测技术,提升无菌保障水平
• 一次性除菌过滤和一次性灌装系统设计应用,有效防止交叉污染
|
| 原材料 |
人员 |
质量体系 |
• 宿主细胞来源清晰,完整的溯源证据链
• 细胞建库培养阶段不使用任何抗生素
• 细胞培养使用化学成分界定的培养基(AOF/无血清)
• 生产过程全程使用无动物源性原料(AOF /无TSE/BSE)
• 药用级辅料和包材
• 原材料和供应商分级管理,定期审计及质量协议确保供应质量保障
|
• 关键人员20+年生物行业生产质量管理经验,50+人生产团队,超百人质量团队
• 年度健康/传染病体检
• 人员卫生培训
• 洁净间权限控制
• 关键操作间人数控制
• 人员更衣资质及无菌操作资质确认
|
• 质量体系符合EMA/US FDA/NMPA GMP以及ISO 9001/ISO 13485
• 采用DMS文档管理系统进行自动化、智能化管理
• 偏差、变更、CAPA、风险管理,实现质量体系持续提升
• 物料、中间品、原液、成品放行管理
• 贯穿产品整个生命周期的验证管理体系(工艺、分析方法、计算机化系统、APS、运输、设备设施、公用系统、仓储、包材密封性)
• 符合FDA 21CFR Part 11要求的计算机化管理平台
• 符合ALCOA+要求的数据完整性管控策略
|
| 质检 |
技术转移 |
仓储和物流 |
• 外源因子检测:无菌,内毒素,支原体,外源病毒等
• 无菌检测:符合Ch.P<1101>和USP<71>
• 内毒素检测:符合Ch.P<1143>和USP<85>
• 支原体检测:符合Ch.P<3301>和USP<63>
• 工艺残留检测:HCP,HCD等
• HCP检测:符合Ch.P<3412>和USP<1103>
• HCD检测:符合Ch.P<3407>和USP<509>
• 部分产品进行动物体内毒性测试:异常毒性,急性毒性评价实验
• 参照 ICH Q2和中美药典进行分析方法验证,确保分析方法准确可靠
• 全面的产品稳定性验证(加速稳定性、长期稳定性、冻融稳定性)
• 仪器设备系统验证及审计追踪管理符合21CFR Part 11
|
• 遵循 WHO《药物生产技术转移指南》,开展技术转移与项目管理,保障产品转移时的质量稳定与一致性
• 技术转移方输出技术转移资料包(工艺开发报告,工艺规程,质量标准,检定规程,分析方法验证报告,物料清单及质检标准,设备清单,细胞株检测报告,产品稳定性数据等)
• 技术接收方进行新产品引入评估,差距分析和共线风险评估,以控制共线和技术风险
• 技术接收方进行设备设施验证,无菌工艺验证,人员培训,环境确认,工艺规程/批记录生效,分析方法确认等
• 不同生产场地切换进行严格的可比性研究分析
• 技术转移经验丰富:60+人技术转移团队,每年平均转产25+个项目,每年迎接40+客户及第三方审计
|
• 产品包装经权威机构检测符合ISTA 3A-2018(2022)标准要求
• 充足完善的全温控仓储空间及C级独立取样间
• 独立完整的仓储分类分区管理机制和质量体系
• 经验证的温度监控系统(Testo)及物料产品管理系统(SAP)
• 物料安全库存预警及MRP管理机制为生产提供保障
• 唯一码提供对产品全生命周期的管理和追溯
• AI动态产品需求预测保证产品持续稳定供应
• 四大仓储物流中心高效服务全球客户
• 全温控国际知名物流商保障产品全球运输安全
• 专业强大的关务团队保证全球进出口合规高效申报
|

























 Star Ribbon预染蛋白Marker蛋白质标记物是生物研究和药物开发的重要组成部分。无论是用于蛋白质电泳还是western blot,我们的预染色蛋白质标记物帮助您快速确定目标蛋白质的分子量或评估转移效率。Fc受体蛋白治疗性抗体的功效取决于Fab片段及其对目标抗原的结合活性,还取决于Fc片段及其与关键Fc受体的相互作用。因此,在抗体工程中候选物必须针对一系列受体进行测试。探索我们的重组Fc受体蛋白质的全面收藏!
Star Ribbon预染蛋白Marker蛋白质标记物是生物研究和药物开发的重要组成部分。无论是用于蛋白质电泳还是western blot,我们的预染色蛋白质标记物帮助您快速确定目标蛋白质的分子量或评估转移效率。Fc受体蛋白治疗性抗体的功效取决于Fab片段及其对目标抗原的结合活性,还取决于Fc片段及其与关键Fc受体的相互作用。因此,在抗体工程中候选物必须针对一系列受体进行测试。探索我们的重组Fc受体蛋白质的全面收藏!
























 膜杰作
膜杰作 Star Staining
Star Staining