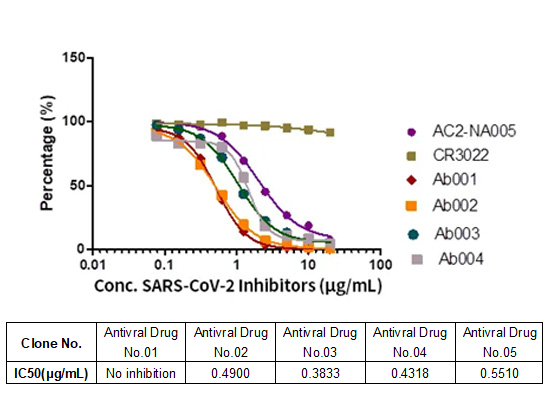
Memory B cell repertoire from triple vaccinees against diverse SARS-CoV-2 variants
Wang K, Jia Z, Bao L, et al.
doi:https://doi.org/10.1038/s41586-022-04466-x
Abstract: Omicron, the most heavily mutated SARS-CoV-2 variant so far, is highly resistant to neutralizing antibodies, raising unprecedented concerns about the effectiveness of antibody therapies and vaccines 1,2. We examined whether sera from individuals who received two or three doses of inactivated vaccine, could neutralize authentic Omicron. The seroconversion rates of neutralizing antibodies were 3.3% (2/60) and 95% (57/60) for 2- and 3-dose vaccinees, respectively. For three-dose recipients, the geometric mean neutralization antibody titre (GMT) of Omicron was 16.5-fold lower than that of the ancestral virus (254). We isolated 323 human monoclonal antibodies (mAbs) derived from memory B cells in 3-dose vaccinees, half of which recognize the receptor binding domain (RBD) and show that a subset of them (24/163) neutralize all SARS-CoV-2 variants of concern (VOCs), including Omicron, potently. Therapeutic treatments with representative broadly neutralizing mAbs were highly protective against SARS-CoV-2 Beta and Omicron infections in mice. Atomic structures of the Omicron Spike in complex with three types of all five VOC-reactive antibodies defined the binding and neutralizing determinants and revealed a key antibody escape site, G446S, that confers greater resistance to one major class of antibodies bound at the right shoulder of RBD through altering local conformation at the binding interface. Our results rationalize the use of 3-dose immunization regimens and suggest that the fundamental epitopes revealed by these broadly ultrapotent antibodies are a rational target for a universal sarbecovirus vaccine.
-
Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron
Zhen Cui, Pan Liu, Nan Wang, et al.
doi:https://doi.org/10.1016/j.cell.2022.01.019
Abstract: The SARS-CoV-2 Omicron variant with increased fitness is spreading rapidly worldwide. Analysis of cryo-EM structures of the spike (S) from Omicron reveals amino acid substitutions forging interactions that stably maintain an active conformation for receptor recognition. The relatively more compact domain organization confers improved stability and enhances attachment but compromises the efficiency of the viral fusion step. Alterations in local conformation, charge, and hydrophobic microenvironments underpin the modulation of the epitopes such that they are not recognized by most NTD- and RBD-antibodies, facilitating viral immune escape. Structure of the Omicron S bound with human ACE2, together with the analysis of sequence conservation in ACE2 binding region of 25 sarbecovirus members, as well as heatmaps of the immunogenic sites and their corresponding mutational frequencies, sheds light on conserved and structurally restrained regions that can be used for the development of broad-spectrum vaccines and therapeutics.
-
Classical and Next-Generation Vaccine Platforms to SARS-CoV-2: Biotechnological Strategies and Genomic Variants
Rachel Siqueira de Queiroz Simões and David Rodríguez-Lázaro
doi:https://doi.org/10.3390/ijerph19042392
Abstract: Several coronaviruses (CoVs) have been identified as human pathogens, including the α-CoVs strains HCoV-229E and HCoV-NL63 and the β-CoVs strains HCoV-HKU1 and HCoV-OC43. SARS-CoV, MERS-CoV, and SARS-CoV-2 are also classified as β-coronavirus. New SARS-CoV-2 spike genomic variants are responsible for human-to-human and interspecies transmissibility, consequences of adaptations of strains from animals to humans. The receptor-binding domain (RBD) of SARS-CoV-2 binds to receptor ACE2 in humans and animal species with high affinity, suggesting there have been adaptive genomic variants. New genomic variants including the incorporation, replacement, or deletion of the amino acids at a variety of positions in the S protein have been documented and are associated with the emergence of new strains adapted to different hosts. Interactions between mutated residues and RBD have been demonstrated by structural modelling of variants including D614G, B.1.1.7, B1.351, P.1, P2; other genomic variants allow escape from antibodies generated by vaccines. Epidemiological and molecular tools are being used for real-time tracking of pathogen evolution and particularly new SARS-CoV-2 variants. COVID-19 vaccines obtained from classical and next-generation vaccine production platforms have entered clinicals trials. Biotechnology strategies of the first generation (attenuated and inactivated virus–CoronaVac, CoVaxin; BBIBPCorV), second generation (replicating-incompetent vector vaccines–ChAdOx-1; Ad5-nCoV; Sputnik V; JNJ-78436735 vaccine-replicating-competent vector, protein subunits, virus-like particles–NVX-CoV2373 vaccine), and third generation (nucleic-acid vaccines–INO-4800 (DNA); mRNA-1273 and BNT 162b (RNA vaccines) have been used. Additionally, dendritic cells (LV-SMENP-DC) and artificial antigen-presenting (aAPC) cells modified with lentiviral vector have also been developed to inhibit viral activity. Recombinant vaccines against COVID-19 are continuously being applied, and new clinical trials have been tested by interchangeability studies of viral vaccines developed by classical and next-generation platforms.
-
The BNT162b2 mRNA SARS-CoV-2 vaccine induces transient afucosylated IgG1 in naive but not antigen-experienced vaccinees
J Van Coillie, T Pongracz, J Rahmoeller, HJ Chen, et.al.
doi: https://doi.org/10.1101/2022.02.14.480353
Abstract: The onset of severe SARS-CoV-2 infection is characterized by the presence of afucosylated IgG1 responses against the viral spike (S) protein, which can trigger exacerbated inflammatory responses. Here, we studied IgG glycosylation after BNT162b2 SARS-CoV-2 mRNA vaccination to explore whether vaccine-induced S protein expression on host cells also generates afucosylated IgG1 responses. SARS-CoV-2 naive individuals initially showed a transient afucosylated anti-S IgG1 response after the first dose, albeit to a lower extent than severely ill COVID-19 patients. In contrast, previously infected, antigen-experienced individuals had low afucosylation levels, which slightly increased after immunization. Afucosylation levels after the first dose correlated with low fucosyltransferase 8 (FUT8) expression levels in a defined plasma cell subset. Remarkably, IgG afucosylation levels after primary vaccination correlated significantly with IgG levels after the second dose. Further studies are needed to assess efficacy, inflammatory potential, and protective capacity of afucosylated IgG responses.
-
Anti-SARS-CoV-2 Omicron Antibodies Isolated from a SARS-CoV-2 Delta Semi-Immune Phage Display Library
I Mendoza-Salazar, KM Gómez-Castellano, et.al.
doi:https://doi.org/10.3390/antib11010013
Abstract: This report describes the discovery and characterization of antibodies with potential broad SARS-CoV-2 neutralization profiles. The antibodies were obtained from a phage display library built with the VH repertoire of a convalescent COVID-19 patient who was infected with SARSCoV-2 B.1.617.2 (Delta). The patient received a single dose of Ad5-nCoV vaccine (Convidecia™, CanSino Biologics Inc.) one month before developing COVID-19 symptoms. Four synthetic VL libraries were used as counterparts of the immune VH repertoire. After three rounds of panning with SARS-CoV-2 receptor-binding domain wildtype (RBD-WT) 34 unique scFvs, were identified, with 27 cross-reactive for the RBD-WT and RBD Delta (RBD-DT), and seven specifics for the RBDWT. The cross-reactive scFvs were more diverse than the RBD-WT specific ones, being encoded by several IGHV genes from the IGHV1 and IGHV3 families combined with short HCDR3s. Six cross-reactive scFvs and one RBD-WT specific scFv were converted to human IgG1 (hIgG1). Out of the seven antibodies, six blocked the RBD-WT binding to angiotensin converting enzyme 2 (ACE2), suggesting these antibodies may neutralize the SARS-CoV-2 infection. Importantly, one of the antibodies also recognized the RBD from the B.1.1.529 (Omicron) isolate, implying that the VH repertoire of the convalescent patient would protect against SARS-CoV-2 Wildtype, Delta, and Omicron. From a practical viewpoint, the triple cross-reactive antibody provides the substrate for developing therapeutic antibodies with a broad SARS-CoV-2 neutralization profile.
-
First identification of SARS-CoV-2 Lambda (C.37) variant in Southern Brazil
Priscila Lamb Wink PhD, Fabiana Caroline Zempulski Volpato MSc, Francielle Liz Monteiro PhD, et.al
doi:https://doi.org/10.1101/2021.06.21.21259241
Abstract: In June 15, 2021, the lineage Lambda (C.37) of SARS-CoV-2 was considered a variant
of interest (VOI) by the World Health Organization. This lineage has high prevalence in some South America countries but it was described only occasionally in Brazil. Here we describe the first report of the SARS-CoV-2 Lambda variant in Southern Brazil. The sequence described in this paper presented all the eight C.37 defining lineage mutations (ORF1a gene: Δ3675-3677; Spike gene: Δ246-252, G75V, T76I, L452Q, F490S, D614G, and T859N) in addition to other 19 mutations. Considering that this VOI has been associated with high rates of transmissibility, the possible spread in the Southern Brazilian community is a matter of concern.
-
Infectivity and immune escape of the new SARS-CoV-2 variant of interest Lambda
Mónica L. Acevedo, Luis Alonso-Palomares, Andrés Bustamante, et.al
doi:https://doi.org/10.1101/2021.06.28.21259673
Background: The newly described SARS-CoV-2 lineage C.37 was recently classified as a variant of interest by the WHO (Lambda variant) based on its high circulation rates in South American countries and the presence of critical mutations in the spike protein. The impact of such mutations in infectivity and immune escape from neutralizing antibodies are entirely unknown.
Methods: We performed a pseudotyped virus neutralization assay and determined the impact of the Lambda variant on infectivity and immune escape using plasma samples from healthcare workers (HCW) from two centers in Santiago, Chile who received the two-doses scheme of the inactivated virus vaccine CoronaVac.
Results: We observed an increased infectivity mediated by the Lambda spike protein that was even higher than that of the D614G (lineage B) or the Alpha and Gamma variants. Compared to the Wild type (lineage A), neutralization was decreased by 3.05-fold for the Lambda variant while it was 2.33-fold for the Gamma variant and 2.03-fold for the Alpha variant.
Conclusions: Our results indicate that mutations present in the spike protein of the Lambda variant of interest confer increased infectivity and immune escape from neutralizing antibodies elicited by CoronaVac. These data reinforce the idea that massive vaccination campaigns in countries with high SARS-CoV-2 circulation must be accompanied by strict genomic surveillance allowing the identification of new isolates carrying spike mutations and immunology studies aimed to determine the impact of these mutations in immune escape and vaccines breakthrough.
-
SARS-CoV-2 Lambda variant exhibits higher infectivity and immune resistance
Izumi Kimura, Yusuke Kosugi, Jiaqi Wu, et.al
doi:https://doi.org/10.1101/2021.07.28.454085
Abstract: SARS-CoV-2 Lambda, a new variant of interest, is now spreading in some South American countries; however, its virological features and evolutionary trait remain unknown. Here we reveal that the spike protein of the Lambda variant is more infectious and it is attributed to the T76I and L452Q mutations. The RSYLTPGD246-253N mutation, a unique 7-amino-acid deletion mutation in the N-terminal domain of the Lambda spike protein, is responsible for evasion from neutralizing antibodies. Since the Lambda variant has dominantly spread according to the increasing frequency of the isolates harboring the RSYLTPGD246-253N mutation, our data suggest that the insertion of the RSYLTPGD246-253N mutation is closely associated with the massive infection spread of the Lambda variant in South America.
-
SARS-CoV-2 Lambda Variant Remains Susceptible to Neutralization by mRNA Vaccine-elicited Antibodies and Convalescent Serum
Takuya Tada, Hao Zhou, Belinda M. Dcosta, et.al
doi:https://doi.org/10.1101/2021.07.02.450959
Abstract: The SARS-CoV-2 lambda variant (lineage C.37) was designated by the World Health Organization as a variant of interest and is currently increasing in prevalence in South American and other countries. The lambda spike protein contains novel mutations within the receptor binding domain (L452Q and F490S) that may contribute to its increased transmissibility and could result in susceptibility to re-infection or a reduction in protection provided by current vaccines. In this study, the infectivity and susceptibility of viruses with the lambda variant spike protein to neutralization by convalescent sera and vaccine elicited antibodies was tested. Virus with the lambda spike had higher infectivity and was neutralized by convalescent sera and vaccine-elicited antibodies with a relatively minor 2.3-3.3-fold decrease in titer on average. The virus was neutralized by the Regeneron therapeutic monoclonal antibody cocktail with no loss of titer. The results suggest that vaccines in current use will remain protective against the lambda variant and that monoclonal antibody therapy will remain effective.
-
Surveillance of SARS-CoV-2 variants in Argentina: detection of Alpha, Gamma, Lambda, Epsilon and Zeta in locally transmitted and imported cases
Torres Carolina, Mojsiejczuk Laura, Acuña Dolores, et.al
doi:https://doi.org/10.1101/2021.07.19.21260779
Abstract: Molecular surveillance of SARS-CoV-2 variants was performed on a total of 2,406 samples from the capital city and nine provinces of Argentina, during 30 epidemiological weeks (EW) that covered the end of the first wave and the beginning of the ongoing second wave of the COVID-19 pandemic in the country (EW 44/2020 to EW 20/2021). The surveillance strategy was mainly based on Sanger sequencing of a Spike coding region that allows the simultaneous identification of signature mutations associated with worldwide circulating variants. In addition, whole SARS-CoV-2 genome sequences were obtained from 456 samples. The main variants found were Gamma, Lambda and Alpha, and to a lesser extent, Zeta and Epsilon. Whereas Gamma dominated in different regions of the country, both Gamma and Lambda prevailed in the most populated area, the metropolitan region of Buenos Aires (MABA), although showing a heterogeneous distribution along this region. This cost-effective surveillance protocol allowed for a rapid response in a limited access to resources scenario, added information on the expansion of the Lambda variant in South America and contributed to the implementation of public health measures to control the disease spread in Argentina.
-
THE EMERGENCE OF SARS-COV-2 VARIANT LAMBDA (C.37) IN SOUTH AMERICA
Pedro E. Romeroa, Alejandra Dávila-Barclay, Guillermo Salvatierra, et.al
doi:https://doi.org/10.1101/2021.06.26.21259487
Abstract: We report the emergence of a novel lineage of SARS-CoV-2 in South America, termed C.37. It presents seven nonsynonymous mutations in the Spike gene (Δ247-253, G75V, T76I, L452Q, F490S, T859N) and a deletion in the ORF1a gene (Δ3675-3677) also found in variants of concern (VOCs) Alpha, Beta, and Gamma. Initially reported in Lima, Peru, in late December 2020, it now accounts for 97% of Peruvian public genomes in April 2021. It is expanding in Chile and Argentina, and there is evidence of onward transmission in Colombia, Ecuador, Mexico, the USA, Germany, and Israel. On June 15, 2021, the World Health Organization designated C.37 as Variant of Interest (VOI) Lambda.
-
Rapid displacement of SARS-CoV-2 variant B.1.1.7 by B.1.617.2 and P.1 in the United States
Alexandre Bolze, Elizabeth T. Cirulli, Shishi Luo, et.al
doi: https://doi.org/10.1101/2021.06.20.21259195
The SARS-CoV-2 variant of concern B.1.617.2 displaced B.1.1.7 as the dominant variant in England and other countries. This study aimed to determine whether B.1.617.2 was also displacing B.1.1.7 in the United States. We analyzed PCR testing results and viral sequencing results of samples collected across the United States, and showed that B.1.1.7 was rapidly being displaced and is no longer responsible for the majority of new cases. The percentage of SARS-CoV-2 positive cases that are B.1.1.7 dropped from 70% in April 2021 to 42% in just 6 weeks. Our analysis showed rapid growth of variants B.1.617.2 and P.1 as the primary drivers for this displacement. Currently, the growth rate of B.1.617.2 was higher than P.1 in the US (0.61 vs. 0.22), which is consistent with reports from other countries. Lastly, we showed that B.1.617.2 was growing faster in counties with a lower vaccination rate.
-
Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum
Chang Liu, Helen M. Ginn, Wanwisa Dejnirattisai, Piyada Supasa, Beibei Wang, et.al
doi: https://doi.org/10.1016/j.cell.2021.06.020
SARS-CoV-2 has undergone progressive change with variants conferring advantage rapidly becoming dominant lineages e.g. B.1.617. With apparent increased transmissibility variant B.1.617.2 has contributed to the current wave of infection ravaging the Indian subcontinent and has been designated a variant of concern in the UK. Here we study the ability of monoclonal antibodies, convalescent and vaccine sera to neutralize B.1.617.1 and B.1.617.2 and complement this with structural analyses of Fab/RBD complexes and map the antigenic space of current variants. Neutralization of both viruses is reduced when compared with ancestral Wuhan related strains but there is no evidence of widespread antibody escape as seen with B.1.351. However, B.1.351 and P.1 sera showed markedly more reduction in neutralization of B.1.617.2 suggesting that individuals previously infected by these variants may be more susceptible to reinfection by B.1.617.2. This observation provides important new insight for immunisation policy with future variant vaccines in non-immune populations.
-
SARS-CoV-2 variant B.1.617 is resistant to Bamlanivimab and evades antibodies induced by infection and vaccination
Markus Hoffmann, Heike Hofmann-Winkler, Nadine Krüger, et.al
doi: https://doi.org/10.1101/2021.05.04.442663
Abstract: The emergence of SARS-CoV-2 variants threatens efforts to contain the COVID-19 pandemic. The number of COVID-19 cases and deaths in India has risen steeply in recent weeks and a novel SARS-CoV-2 variant, B.1.617, is believed to be responsible for many of these cases. The spike protein of B.1.617 harbors two mutations in the receptor binding domain, which interacts with the ACE2 receptor and constitutes the main target of neutralizing antibodies. Therefore, we analyzed whether B.1.617 is more adept in entering cells and/or evades antibody responses. B.1.617 entered two out of eight cell lines tested with slightly increased efficiency and was blocked by entry inhibitors. In contrast, B.1.617 was resistant against Bamlanivimab, an antibody used for COVID-19 treatment. Finally, B.1.617 evaded antibodies induced by infection or vaccination, although with moderate efficiency. Collectively, our study reveals that antibody evasion of B.1.617 may contribute to the rapid spread of this variant.
-
SARS-CoV-2 variant B.1.617 is resistant to Bamlanivimab and evades antibodies induced by infection and vaccination
Markus Hoffmann, Heike Hofmann-Winkler, Nadine Krüger, et.al
doi: https://doi.org/10.1101/2021.05.04.442663
Abstract: The emergence of SARS-CoV-2 variants threatens efforts to contain the COVID-19 pandemic. The number of COVID-19 cases and deaths in India has risen steeply in recent weeks and a novel SARS-CoV-2 variant, B.1.617, is believed to be responsible for many of these cases. The spike protein of B.1.617 harbors two mutations in the receptor binding domain, which interacts with the ACE2 receptor and constitutes the main target of neutralizing antibodies. Therefore, we analyzed whether B.1.617 is more adept in entering cells and/or evades antibody responses. B.1.617 entered two out of eight cell lines tested with slightly increased efficiency and was blocked by entry inhibitors. In contrast, B.1.617 was resistant against Bamlanivimab, an antibody used for COVID-19 treatment. Finally, B.1.617 evaded antibodies induced by infection or vaccination, although with moderate efficiency. Collectively, our study reveals that antibody evasion of B.1.617 may contribute to the rapid spread of this variant.
-
Possible link between higher transmissibility of B.1.617 and B.1.1.7 variants of SARS-CoV-2 and increased structural stability of its spike protein and hACE2 affinity
Vipul Kumar1, Jasdeep Singh, Seyed E. Hasnain, Durai Sundar
doi: https://doi.org/10.1101/2021.04.29.441933
Abstract: The Severe Acute syndrome corona Virus 2 (SARS-CoV-2) outbreak in December 2019 has caused a global pandemic. The rapid mutation rate in the virus has caused alarming situations worldwide and is being attributed to the false negativity in RT-PCR tests, which also might lead to inefficacy of the available drugs. It has also increased the chances of reinfection and immune escape. We have performed Molecular Dynamic simulations of three different Spike-ACE2 complexes, namely Wildtype (WT), B.1.1.7 variant (N501Y Spike mutant) and B.1.617 variant (L452R, E484Q Spike mutant) and compared their dynamics, binding energy and molecular interactions. Our result shows that mutation has caused the increase in the binding energy between the Spike and hACE2. In the case of B.1.617 variant, the mutations at L452R and E484Q increased the stability and intra-chain interactions in the Spike protein, which may change the interaction ability of human antibodies to this Spike variant. Further, we found that the B.1.1.7 variant had increased hydrogen interaction with LYS353 of hACE2 and more binding affinity in comparison to WT. The current study provides the biophysical basis for understanding the molecular mechanism and rationale behind the increase in the transmissivity and infectivity of the mutants compared to wild-type SARS-CoV-2.
-
Neutralization of variant under investigation B.1.617 with sera of BBV152 vaccinees
Pragya D. Yadav, Gajanan N. Sapkal, Priya Abraham, M.D
doi: https://doi.org/10.1101/2021.04.23.441101
Abstract: The drastic rise in the number of cases in Maharashtra, India has created a matter of concern for public health experts. Twelve isolates of VUI lineage B.1.617 were propagated in VeroCCL81 cells and characterized. Convalescent sera of the COVID-19 cases and recipients of BBV152 (Covaxin) were able to neutralize VUI B.1.617.
-
Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India
Sarah Cherian, Varsha Potdar, Santosh Jadhav, et al.
doi: https://doi.org/10.1101/2021.04.22.440932
Abstract: As the global severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic expands, genomic epidemiology and whole genome sequencing are being constantly used to investigate its transmissions and evolution. In the backdrop of the global emergence of “variants of concern” (VOCs) during December 2020 and an upsurge in a state in the western part of India since January 2021, whole genome sequencing and analysis of spike protein mutations using sequence and structural approaches was undertaken to identify possible new variants and gauge the fitness of current circulating strains. Phylogenetic analysis revealed that the predominant clade in circulation was a distinct newly identified lineage B.1.617 possessing common signature mutations D111D, G142D, L452R, E484Q, D614G and P681R, in the spike protein including within the receptor binding domain (RBD). Of these, the mutations at residue positions 452, 484 and 681 have been reported in other globally circulating lineages. The structural analysis of RBD mutations L452R and E484Q along with P681R in the furin cleavage site, may possibly result in increased ACE2 binding and rate of S1-S2 cleavage resulting in better transmissibility. The same two RBD mutations indicated decreased binding to selected monoclonal antibodies (mAbs) and may affect their neutralization potential. Experimental validation is warranted for accessing both ACE2 binding and the effectiveness of commonly elicited neutralizing mAbs for the strains of lineage B.1.617. The emergence of such local variants through the accumulation of convergent mutations during the COVID-19 second wave needs to be further investigated for their public health impact in the rest of the country and its possibility of becoming a VOC.
-
Bioinformatics analysis of SARS-CoV-2 RBD mutant variants and insights into antibody and ACE2 receptor binding
Prashant Ranjan, Neha, Chandra Devi1and Parimal Das
doi: https://doi.org/10.1101/2021.04.03.438113
Abstract: Prevailing COVID-19 vaccines are based on the spike protein of earlier SARS-CoV-2 strain that emerged in Wuhan, China. Continuously evolving nature of SARS-CoV-2 resulting emergence of new variant/s raise the risk of immune absconds. Several RBD (receptor-binding domain) variants have been reported to affect the vaccine efficacy considerably. In the present study, we performed in silico structural analysis of spike protein of double mutant (L452R & E484Q), a new variant of SARS-CoV-2 recently reported in India along with K417G variants and earlier reported RBD variants and found structural changes in RBD region after comparing with the wild type. Comparison of the binding affinity of the double mutant and earlier reported RBD variant for ACE2 (angiotensin 2 altered enzymes) receptor and CR3022 antibody with the wildtype strain revealed the lowest binding affinity of the double mutant for CR3022 among all other variants. These findings suggest that the newly emerged double mutant could significantly reduce the impact of the current vaccine which threatens the protective efficacy of current vaccine therapy.
-
Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7
Pengfei Wang, Manoj S. Nair, Lihong Liu et al
doi: https://doi.org/10.1101/2021.01.25.428137
Abstract: The COVID-19 pandemic has ravaged the globe, and its causative agent, SARS-CoV-2, continues to rage. Prospects of ending this pandemic rest on the development of effective interventions. Single and combination monoclonal antibody(mAb) therapeutics have received emergency use authorization, with more in the pipeline. Furthermore, multiple vaccine constructs have shown promise8, including two with ~95% protective efficacy against COVID-19. However, these interventions were directed toward the initial SARS-CoV-2 that emerged in 2019. The recent emergence of new SARS-CoV-2 variants B.1.1.7 in the UK and B.1.351 in South Africa is of concern because of their purported ease of transmission and extensive mutations in the spike protein. We now report that B.1.1.7 is refractory to neutralization by most mAbs to the N-terminal domain (NTD) of spike and relatively resistant to a few mAbs to the receptor-binding domain (RBD). It is not more resistant to convalescent plasma or vaccinee sera. Findings on B.1.351 are more worrisome in that this variant is not only refractory to neutralization by most NTD mAbs but also by multiple individual mAbs to the receptor-binding motif on RBD, largely due to an E484K mutation. Moreover, B.1.351 is markedly more resistant to neutralization by convalescent plasma (9.4 fold) and vaccinee sera (10.3-12.4 fold). B.1.351 and emergent variants with similar spike mutations present new challenges for mAb therapy and threaten the protective efficacy of current vaccines.
-
SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma
Constantinos Kurt Wibmer, Frances Ayres, Tandile Hermanus et al
doi: https://doi.org/10.1101/2021.01.18.427166
Abstract: SARS-CoV-2 501Y.V2, a novel lineage of the coronavirus causing COVID-19, contains multiple mutations within two immunodominant domains of the spike protein. Here we show that this lineage exhibits complete escape from three classes of therapeutically relevant monoclonal antibodies. Furthermore 501Y.V2 shows substantial or complete escape from neutralizing antibodies in COVID-19 convalescent plasma. These data highlight the prospect of reinfection with antigenically distinct variants and may foreshadow reduced efficacy of current spike-based vaccines.
-
Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies
Allison J. Greaney, Andrea N. Loes, Katharine H.D. Crawford et al
doi: https://doi.org/10.1101/2020.12.31.425021
Abstract: The evolution of SARS-CoV-2 could impair recognition of the virus by human antibody-mediated immunity. To facilitate prospective surveillance for such evolution, we map how convalescent serum antibodies are impacted by all mutations to the spike’s receptor-binding domain (RBD), the main target of serum neutralizing activity. Binding by polyclonal serum antibodies is affected by mutations in three main epitopes in the RBD, but there is substantial variation in the impact of mutations both among individuals and within the same individual over time. Despite this inter- and intra-person heterogeneity, the mutations that most reduce antibody binding usually occur at just a few sites in the RBD’s receptor binding motif. The most important site is E484, where neutralization by some sera is reduced >10-fold by several mutations, including one in emerging viral lineages in South Africa and Brazil. Going forward, these serum escape maps can inform surveillance of SARS-CoV-2 evolution.
-
Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data
Volz, Mishra*, Chand et al
doi: https://doi.org/10.1101/2020.12.30.20249034
Abstract: The SARS-CoV-2 lineage B.1.1.7, now designated Variant of Concern 202012/01 (VOC) by Public Health England, originated in the UK in late Summer to early Autumn 2020. We examine epidemiological evidence for this VOC having a transmission advantage from several perspectives. First, whole genome sequence data collected from community-based diagnostic testing provides an indication of changing prevalence of different genetic variants through time. Phylodynamic modelling additionally indicates that genetic diversity of this lineage has changed in a manner consistent with exponential growth. Second, we find that changes in VOC frequency inferred from genetic data correspond closely to changes inferred by S-gene target failures (SGTF) in community-based diagnostic PCR testing. Third, we examine growth trends in SGTF and non-SGTF case numbers at local area level across England, and show that the VOC has higher transmissibility than non-VOC lineages, even if the VOC has a different latent period or generation time. Available SGTF data indicate a shift in the age composition of reported cases, with a larger share of under 20 year olds among reported VOC than non-VOC cases. Fourth, we assess the association of VOC frequency with independent estimates of the overall SARS-CoV-2 reproduction number through time. Finally, we fit a semi-mechanistic model directly to local VOC and non-VOC case incidence to estimate the reproduction numbers over time for each. There is a consensus among all analyses that the VOC has a substantial transmission advantage, with the estimated difference in reproduction numbers between VOC and non-VOC ranging between 0.4 and 0.7, and the ratio of reproduction numbers varying between 1.4 and 1.8. We note that these estimates of transmission advantage apply to a period where high levels of social distancing were in place in England; extrapolation to other transmission contexts therefore requires caution.
-
Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England
Davies, Barnard, Jarvis et al
doi: https://doi.org/10.1101/2020.12.24.20248822
Abstract: A novel SARS-CoV-2 variant, VOC 202012/01, emerged in southeast England in November 2020 and appears to be rapidly spreading towards fixation. We fitted a two-strain mathematical model of SARS-CoV-2 transmission to observed COVID-19 hospital admissions, hospital and ICU bed occupancy, and deaths; SARS-CoV-2 PCR prevalence and seroprevalence; and the relative frequency of VOC 202012/01 in the three most heavily affected NHS England regions (South East, East of England, and London). We estimate that VOC 202012/01 is 56% more transmissible (95% credible interval across three regions 50-74%) than preexisting variants of SARS-CoV-2. We were unable to find clear evidence that VOC 202012/01 results in greater or lesser severity of disease than preexisting variants. Nevertheless, the increase in transmissibility is likely to lead to a large increase in incidence, with COVID-19 hospitalisations and deaths projected to reach higher levels in 2021 than were observed in 2020, even if regional tiered restrictions implemented before 19 December are maintained. Our estimates suggest that control measures of a similar stringency to the national lockdown implemented in England in November 2020 are unlikely to reduce the effective reproduction number Rt to less than 1, unless primary schools, secondary schools, and universities are also closed. We project that large resurgences of the virus are likely to occur following easing of control measures. It may be necessary to greatly accelerate vaccine roll-out to have an appreciable impact in suppressing the resulting disease burden.
-
Early empirical assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020
Leung, Shum, Leung et al
doi: https://doi.org/10.1101/2020.12.20.20248581
Abstract: Two new SARS-CoV-2 lineages with the N501Y mutation in the receptor binding domain of the spike protein have rapidly become prevalent in the UK. We estimated that the earlier 501Y lineage without amino acid deletion Δ69/Δ70 circulating mainly between early September to mid-November was 10% (6-13%) more transmissible than the 501N lineage, and the currently dominant 501Y lineage with amino acid deletion Δ69/Δ70 circulating since late September was 75% (70-80%) more transmissible than the 501N lineage.
-
Mutation Landscape of SARS-CoV-2 in Africa
Nassir, Musanabaganwa, Mwikarago
doi: https://doi.org/10.1101/2020.12.20.423630
Abstract: COVID-19 disease has had a relatively less severe impact in Africa. To understand the role of SARS CoV2 mutations on COVID-19 disease in Africa, we analysed 282 complete nucleotide sequences from African isolates deposited in the NCBI Virus Database. Sequences were aligned against the prototype Wuhan sequence (GenBank accession: NC_045512.2) in BWA v. 0.7.17. SAM and BAM files were created, sorted and indexed in SAMtools v. 1.10 and marked for duplicates using Picard v. 2.23.4. Variants were called with mpileup in BCFtools v. 1.11. Phylograms were created using Mr. Bayes v 3.2.6. A total of 2,349 single nucleotide polymorphism (SNP) profiles across 294 sites were identified. Clades associated with severe disease in the United States, France, Italy, and Brazil had low frequencies in Africa (L84S=2.5%, L3606F=1.4%, L3606F/V378I/=0.35, G251V=2%). Sub Saharan Africa (SSA) accounted for only 3% of P323L and 4% of Q57H mutations in Africa. Comparatively low infections in SSA were attributed to the low frequency of the D614G clade in earlier samples (25% vs 67% global). Higher disease burden occurred in countries with higher D614G frequencies (Egypt=98%, Morocco=90%, Tunisia=52%, South Africa) with D614G as the first confirmed case. V367F, D364Y, V483A and G476S mutations associated with efficient ACE2 receptor binding and severe disease were not observed in Africa. 95% of all RdRp mutations were deaminations leading to CpG depletion and possible attenuation of virulence. More genomic and experimental studies are needed to increase our understanding of the temporal evolution of the virus in Africa, clarify our findings, and reveal hot spots that may undermine successful therapeutic and vaccine interventions.
-
Major new lineages of SARS-CoV-2 emerge and spread in South Africa during lockdown
Tegally, Wilkinson, Lessells et al
doi: https://www.medrxiv.org/content/10.1101/2020.10.28.20221143v1
Abstract: In March 2020, the first cases of COVID-19 were reported in South Africa. The epidemic spread very fast despite an early and extreme lockdown and infected over 600,000 people, by far the highest number of infections in an African country. To rapidly understand the spread of SARS-CoV-2 in South Africa, we formed the Network for Genomics Surveillance in South Africa (NGS-SA). Here, we analyze 1,365 high quality whole genomes and identify 16 new lineages of SARS-CoV-2. Most of these unique lineages have mutations that are found hardly anywhere else in the world. We also show that three lineages spread widely in South Africa and contributed to ∼42% of all of the infections in the country. This included the first identified C lineage of SARS-CoV-2, C.1, which has 16 mutations as compared with the original Wuhan sequence. C.1 was the most geographically widespread lineage in South Africa, causing infections in multiple provinces and in all of the eleven districts in KwaZulu-Natal (KZN), the most sampled province. Interestingly, the first South-African specific lineage, B.1.106, which was identified in April 2020, became extinct after nosocomial outbreaks were controlled. Our findings show that genomic surveillance can be implemented on a large scale in Africa to identify and control the spread of SARS-CoV-2.
-
Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa
Tegally, Wilkinson, Giovanetti et al
doi: https://doi.org/10.1101/2020.12.21.20248640
Summary: Continued uncontrolled transmission of the severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) in many parts of the world is creating the conditions for significant virus evolution. Here, we describe a new SARS-CoV-2 lineage (501Y.V2) characterised by eight lineage-defining mutations in the spike protein, including three at important residues in the receptor-binding domain (K417N, E484K and N501Y) that may have functional significance. This lineage emerged in South Africa after the first epidemic wave in a severely affected metropolitan area, Nelson Mandela Bay, located on the coast of the Eastern Cape Province. This lineage spread rapidly, becoming within weeks the dominant lineage in the Eastern Cape and Western Cape Provinces. Whilst the full significance of the mutations is yet to be determined, the genomic data, showing the rapid displacement of other lineages, suggest that this lineage may be associated with increased transmissibility.
-
Early transmission of SARS-CoV-2 in South Africa: An epidemiological and phylogenetic report
Giandharia, Pillaya, Wilkinson et al
Int J Infect Dis(2020)11, 128
doi: https://doi.org/10.1016/j.ijid.2020.11.128
Abstract: Objectives: The Network for Genomic Surveillance in South Africa (NGS-SA) was formed to investigate the introduction and understand the early transmission dynamics of the SARS-CoV-2 epidemic in South-Africa.
Design: This paper presents the first results from this group, which is a molecular epidemiological study of the first 21 SARS-CoV-2 whole genomes sampled in the first port of entry – KwaZulu-Natal (KZN) –during the first month of the epidemic. By combining this with calculations of the effective reproduction number (R), it aimed to shed light on the patterns of infections in South Africa.
Results: Two of the largest provinces – Gauteng and KZN – had a slow growth rate for the number of detected cases, while the epidemic spread faster in the Western Cape and Eastern Cape. The estimates of transmission potential suggested a decrease towards R = 1 since the first cases and deaths, but a subsequent estimated R average of 1.39 between 6–18 May 2020. It was also demonstrated that early transmission in KZN was associated with multiple international introductions and dominated by lineages B1 and B. Evidence for locally acquired infections in a hospital in Durban within the first month of the epidemic was also provided.
Conclusion: The COVID-19 pandemic in South Africa was very heterogeneous in its spatial dimension, with many distinct introductions of SARS-CoV2 in KZN and evidence of nosocomial transmission, which inflated early mortality in KZN. The epidemic at the local level was still developing and NGS-SA aimed to clarify the dynamics in South Africa and devise the most effective measures as the outbreak evolved.
-
Brief report: New Variant Strain of SARS-CoV-2 Identified in Travelers from Brazil
January 12, 2021
National Institute of Infectious Diseases, JAPAN
Technical detail: The variant isolate (GISAID ID: EPI_ISL_792680 to 792683) belongs to B.1.1.248 lineage and has 12 mutations in the spike protein, including N501Y and E484K. - N501Y is a mutation found in variant strains including VOC-202012/01 and 501Y.V2, implicated to increase transmissibility. - The E484K was reported to be an escape mutation from a monoclonal antibody which neutralize SARSCoV-2 (1,2). The E484K has been observed in variant isolates escaping from convalescent plasma (3) and with a 10-fold decrease in neutralization capability by convalescent plasma (4)(both in preprint articles), suggesting possible change in antigenicity. - In Brazil, a variant isolate with E484K belonging to B.1.1.248 was reported on January 6, 2021 (5), but it is not identical to the new variant isolate identified in Japan.
-
Researchers Discover New Variant of COVID-19 Virus in Columbus, Ohio
January 13, 2021
The Ohio State University Wexner Medical Center
Scientists at The Ohio State University Wexner Medical Center and College of Medicine have discovered a new variant of SARS-Cov-2, the virus that causes COVID-19. The new variant carries a mutation identical to the U.K. strain, but it likely arose in a virus strain already present in the United States. The researchers also report the evolution of another U.S. strain that acquired three other gene mutations not previously seen together in SARS-CoV2.







 关注公众号
关注公众号